van der Spoel lab
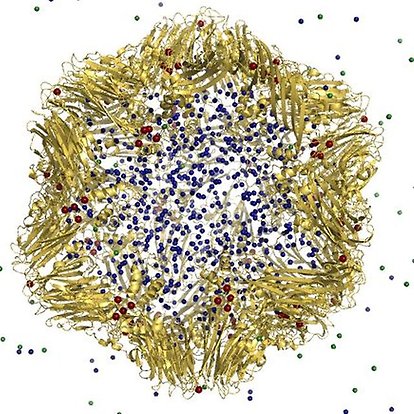
"…Everything that living things do can be understood in terms of the jiggling and wiggling of atoms…" is a fame quote by Richard Feynman. Although it is easier said than done my group is working hard to make it possible to predict the " jiggling and wiggling of atoms" using computer simulation based on accurate physical models. With these and other models we study biological systems, e.g. virus particles as well as partial virus capsids in order to understand virus infection and budding at the molecular level. Finally we are studying how pollutants interfere with biological receptors and DNA.
Popular science presentation
...
Research projects
"…Everything that living things do can be understood in terms of the jiggling and wiggling of atoms…" is a fame quote by Richard Feynman. It is easier said than done however. My group is working hard to make this happen. The long-term goals of my research group are therefore
- to create physical models (force fields) of compounds and molecules that faithfully reproduce the (free) energies of these compounds,
- to implement tools in the GROMACS (http://www.gromacs.org) package for simulations using these models,
- to develop and implement tools for the analysis of structural, dynamic and energetic properties of molecular systems and
- to perform systematic benchmarks of the predictive power of existing force fields and quantum-chemistry theories with the purpose of generating reference systems for the development of better force fields.
Simultaneously we are using traditional as well as more modern force fields to study a range of systems:
- Simulations of entire virus particles as well as partial virus capsids are performed to study virus infection and budding.
- Simulations of RNA in solution and in virus particles.
- Systematic docking and binding calculations are done to understand how pollutants interfere with biological receptors and DNA.
Welcome to our external homepage
Group members
Publications
Martini on the Rocks: Can a Coarse-Grained Force Field Model Crystals?
Part of Journal of Physical Chemistry Letters, p. 1079-1088, 2024
- DOI for Martini on the Rocks: Can a Coarse-Grained Force Field Model Crystals?
- Download full text (pdf) of Martini on the Rocks: Can a Coarse-Grained Force Field Model Crystals?

Part of Journal of Chemical Theory and Computation, p. 2362-2376, 2024
- DOI for Impact of Combination Rules, Level of Theory, and Potential Function on the Modeling of Gas- and Condensed-Phase Properties of Noble Gases
- Download full text (pdf) of Impact of Combination Rules, Level of Theory, and Potential Function on the Modeling of Gas- and Condensed-Phase Properties of Noble Gases
Can molecular dynamics be used to simulate biomolecular recognition?
Part of Journal of Chemical Physics, 2023
Simulations of Amyloid-Forming Peptides in the Crystal State
Part of The Protein Journal, p. 192-204, 2023
- DOI for Simulations of Amyloid-Forming Peptides in the Crystal State
- Download full text (pdf) of Simulations of Amyloid-Forming Peptides in the Crystal State
Probing Phase Transitions in Organic Crystals Using Atomistic MD Simulations
Part of ACS PHYSICAL CHEMISTRY AU, p. 84-93, 2023
- DOI for Probing Phase Transitions in Organic Crystals Using Atomistic MD Simulations
- Download full text (pdf) of Probing Phase Transitions in Organic Crystals Using Atomistic MD Simulations
Binding Networks Identify Targetable Protein Pockets for Mechanism-Based Drug Design
Part of International Journal of Molecular Sciences, 2022
- DOI for Binding Networks Identify Targetable Protein Pockets for Mechanism-Based Drug Design
- Download full text (pdf) of Binding Networks Identify Targetable Protein Pockets for Mechanism-Based Drug Design
Editorial overview: Theory and simulation and their new friends
Part of Current opinion in structural biology, 2021
Part of Journal of Chemical Physics, 2021
Part of Journal of Chemical Physics, 2021
Accurate absolute free energies for ligand-protein binding based on non-equilibrium approaches
Part of Communications Chemistry, 2021
- DOI for Accurate absolute free energies for ligand-protein binding based on non-equilibrium approaches
- Download full text (pdf) of Accurate absolute free energies for ligand-protein binding based on non-equilibrium approaches
Microscopic origins of conductivity in molten salts unraveled by computer simulations
Part of Communications Chemistry, 2021
- DOI for Microscopic origins of conductivity in molten salts unraveled by computer simulations
- Download full text (pdf) of Microscopic origins of conductivity in molten salts unraveled by computer simulations
NMR Refinement and Peptide Folding Using the GROMACS Software
Part of Journal of Biomolecular NMR, p. 143-149, 2021
- DOI for NMR Refinement and Peptide Folding Using the GROMACS Software
- Download full text (pdf) of NMR Refinement and Peptide Folding Using the GROMACS Software
Making Soup: Preparing and Validating Models of the Bacterial Cytoplasm for Molecular Simulation
Part of Journal of Chemical Information and Modeling, p. 322-331, 2020
Theoretical Infrared Spectra: Quantitative Similarity Measures and Force Fields
Part of Journal of Chemical Theory and Computation, p. 3307-3315, 2020
- DOI for Theoretical Infrared Spectra: Quantitative Similarity Measures and Force Fields
- Download full text (pdf) of Theoretical Infrared Spectra: Quantitative Similarity Measures and Force Fields
An Intuitively Understandable Quality Measure for Theoretical Vibrational Spectra
Part of Journal of Physical Chemistry Letters, p. 5471-5475, 2020
- DOI for An Intuitively Understandable Quality Measure for Theoretical Vibrational Spectra
- Download full text (pdf) of An Intuitively Understandable Quality Measure for Theoretical Vibrational Spectra
A potential for molecular simulation of compounds with linear moieties
Part of Journal of Chemical Physics, 2020
- DOI for A potential for molecular simulation of compounds with linear moieties
- Download full text (pdf) of A potential for molecular simulation of compounds with linear moieties
Toward a Computational Ecotoxicity Assay
Part of Journal of Chemical Information and Modeling, p. 3792-3803, 2020
Propagation of uncertainty in physicochemical data to force field predictions
Part of Physical Review Research, 2020
- DOI for Propagation of uncertainty in physicochemical data to force field predictions
- Download full text (pdf) of Propagation of uncertainty in physicochemical data to force field predictions
Part of PloS Computational Biology, 2019
- DOI for Ten simple rules on how to create open access and reproducible molecular simulations of biological systems
- Download full text (pdf) of Ten simple rules on how to create open access and reproducible molecular simulations of biological systems
Role of Host-Guest Charge Transfer in Cyclodextrin Complexation: A Computational Study
Part of The Journal of Physical Chemistry C, p. 17745-17756, 2019
Molten alkali halides - temperature dependence of structure, dynamics and thermodynamics
Part of Physical Chemistry, Chemical Physics - PCCP, p. 18516-18524, 2019
Direct Link between Structure, Dynamics, and Thermodynamics in Molten Salts
Part of The Journal of Physical Chemistry C, p. 25596-25602, 2019
Systematically improved melting point prediction: a detailed physical simulation model is required
Part of Chemical Communications, p. 12044-12047, 2019
- DOI for Systematically improved melting point prediction: a detailed physical simulation model is required
- Download full text (pdf) of Systematically improved melting point prediction: a detailed physical simulation model is required
Rotational and Translational Diffusion of Proteins as a Function of Concentration
Part of ACS Omega, p. 20654-20664, 2019
- DOI for Rotational and Translational Diffusion of Proteins as a Function of Concentration
- Download full text (pdf) of Rotational and Translational Diffusion of Proteins as a Function of Concentration
Prediction of Partition Coefficients of Environmental Toxins Using Computational Chemistry Methods
Part of ACS Omega, p. 13772-13781, 2019
- DOI for Prediction of Partition Coefficients of Environmental Toxins Using Computational Chemistry Methods
- Download full text (pdf) of Prediction of Partition Coefficients of Environmental Toxins Using Computational Chemistry Methods
Transient isomers in the photodissociation of bromoiodomethane
Part of Journal of Chemical Physics, 2018
Force Field Benchmark of Amino Acids: I. Hydration and Diffusion in Different Water Models
Part of Journal of Chemical Information and Modeling, p. 1037-1052, 2018
Statistical efficiency of methods for computing free energy of hydration
Part of Journal of Chemical Physics, 2018
- DOI for Statistical efficiency of methods for computing free energy of hydration
- Download full text (pdf) of Statistical efficiency of methods for computing free energy of hydration
Impact of Dispersion Coefficient on Simulations of Proteins and Organic Liquids
Part of Journal of Physical Chemistry B, p. 8018-8027, 2018
Small Molecule Thermochemistry: A Tool for Empirical Force Field Development
Part of Journal of Physical Chemistry A, p. 8982-8988, 2018
Part of Scientific Data, 2018
- DOI for The Alexandria library, a quantum-chemical database of molecular properties for force field development
- Download full text (pdf) of The Alexandria library, a quantum-chemical database of molecular properties for force field development
Phase-Transferable Force Field for Alkali Halides
Part of Journal of Chemical Theory and Computation, p. 5933-5948, 2018
Part of Journal of Chemical Theory and Computation, p. 5553-5566, 2018
Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations
Part of Nucleic Acids Research, p. 4872-4882, 2018
- DOI for Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations
- Download full text (pdf) of Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations
Part of Journal of Physical Chemistry Letters, p. 2705-2712, 2017
Part of Journal of Chemical Theory and Computation, p. 1034-1043, 2017
Part of Journal of Physics, 2017
Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1
Part of Molecular Cell, p. 847-8590000000, 2017
- DOI for Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1
- Download full text (pdf) of Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1
Systematic exploration of multiple drug binding sites
Part of Journal of Cheminformatics, 2017
- DOI for Systematic exploration of multiple drug binding sites
- Download full text (pdf) of Systematic exploration of multiple drug binding sites
Part of Journal of Chemical Physics, 2016
- DOI for Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity
- Download full text (pdf) of Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity
Atomistic Simulation of Protein Encapsulation in Metal-Organic Frameworks
Part of Journal of Physical Chemistry B, p. 477-484, 2016
Exploration of Interfacial Hydration Networks of Target Ligand Complexes
Part of Journal of Chemical Information and Modeling, p. 148-158, 2016
Part of Journal of Chemical Information and Modeling, p. 2080-2092, 2016
- DOI for Evaluation of Generalized Born Models for Large Scale Affinity Prediction of Cyclodextrin Host-Guest Complexes
- Download full text (pdf) of Evaluation of Generalized Born Models for Large Scale Affinity Prediction of Cyclodextrin Host-Guest Complexes
Binding of Pollutants to Biomolecules: A Simulation Study
Part of Chemical Research in Toxicology, p. 1679-1688, 2016
- DOI for Binding of Pollutants to Biomolecules: A Simulation Study
- Download full text (pdf) of Binding of Pollutants to Biomolecules: A Simulation Study
Part of Journal of Chemical Physics, 2016
Part of Journal of Chemical Theory and Computation, p. 5103-5113, 2015
Properties of Organic Liquids when Simulated with Long-Range Lennard-Jones Interactions
Part of Journal of Chemical Theory and Computation, p. 2938-2944, 2015
Part of Journal of Computational Chemistry, p. 1473-1479, 2015
Part of Journal of Molecular Structure, p. 196-202, 2015
Mobility-based prediction of hydration structures of protein surfaces
Part of Bioinformatics, p. 1959-1965, 2015
Deciphering Solution Scattering Data with Experimentally Guided Molecular Dynamics Simulations
Part of Journal of Chemical Theory and Computation, p. 780-787, 2015
- DOI for Deciphering Solution Scattering Data with Experimentally Guided Molecular Dynamics Simulations
- Download full text (pdf) of Deciphering Solution Scattering Data with Experimentally Guided Molecular Dynamics Simulations
Force Field Benchmark of Organic Liquids. 2. Gibbs Energy of Solvation
Part of JOURNAL OF CHEMICAL INFORMATION AND MODELING, p. 1192-1201, 2015
Part of PLOS ONE, 2015
- DOI for Deconvoluting Protein (Un)folding Structural Ensembles Using X-Ray Scattering, Nuclear Magnetic Resonance Spectroscopy and Molecular Dynamics Simulation
- Download full text (pdf) of Deconvoluting Protein (Un)folding Structural Ensembles Using X-Ray Scattering, Nuclear Magnetic Resonance Spectroscopy and Molecular Dynamics Simulation
Thermodynamics of hydronium and hydroxide surface solvation
Part of Chemical Science, p. 1745-1749, 2014
Part of Journal of Chemical Theory and Computation, p. 5606-5615, 2014
Part of The Journal of Physical Chemistry C, p. 7163-7173, 2014
CO2 and O-2 Distribution in Rubisco Suggests the Small Subunit Functions as a CO2 Reservoir
Part of Journal of the American Chemical Society, p. 3165-3171, 2014
Thiamin Function, Metabolism, Uptake, and Transport
Part of Biochemistry, p. 821-835, 2014
Part of Bioinformatics, p. 439-441, 2014
Part of Journal of Computational Chemistry, p. 260-269, 2014
Local Partition Coefficients Govern Solute Permeability of Cholesterol-Containing Membranes
Part of Biophysical Journal, p. 2760-2770, 2013
Transcription-factor binding and sliding on DNA studied using micro- and macroscopic models
Part of Proceedings of the National Academy of Sciences of the United States of America, p. 19796-19801, 2013
Part of Environmental Science and Technology, p. 7421-7429, 2013
Part of Peptides, p. 94-100, 2013
GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit
Part of Bioinformatics, p. 845-854, 2013
Unexpected Effects of Cholesterol on Membrane Permeability
Part of Biophysical Journal, 2013
Part of Biochemistry, p. 7798-7806, 2013
Quantification of Solvent Contribution to the Stability of Noncovalent Complexes
Part of Journal of Chemical Theory and Computation, p. 4542-4551, 2013
Organic molecules on the surface of water droplets: an energetic perspective
Part of Physical Chemistry, Chemical Physics - PCCP, p. 9537-9545, 2012
GROMACS molecule & liquid database
Part of Bioinformatics, p. 752-753, 2012
Part of Journal of the American Society for Mass Spectrometry, p. 1319-1325, 2012
Molecular recognition in different environments: β-Cyclodextrin dimer formation in organic solvents
Part of Journal of Physical Chemistry B, p. 12684-12693, 2012
Free Energy of Separation of Structure II Clathrate Hydrate in Water and a Light Oil
Part of Journal of Physical Chemistry B, p. 5933-5940, 2012
Part of Journal of Chemical Theory and Computation, p. 2474-2483, 2012
Virus Capsid Dissolution Studied by Microsecond Molecular Dynamics Simulations
Part of PloS Computational Biology, 2012
- DOI for Virus Capsid Dissolution Studied by Microsecond Molecular Dynamics Simulations
- Download full text (pdf) of Virus Capsid Dissolution Studied by Microsecond Molecular Dynamics Simulations
Large Influence of Cholesterol on Solute Partitioning into Lipid Membranes
Part of Journal of the American Chemical Society, p. 5351-5361, 2012
Part of Journal of Chemical Theory and Computation, p. 61-74, 2012
Part of Journal of Physical Chemistry B, p. 3880-3889, 2012
Part of Wiley Interdisciplinary Reviews: Computational Molecular Science, p. 710-715, 2011
Part of Physical Review E. Statistical, Nonlinear, and Soft Matter Physics, 2011
Part of Journal of Molecular Modeling, p. 3289-3297, 2011
Trajectory NG: portable, compressed, general molecular dynamics trajectories
Part of Journal of Molecular Modeling, p. 2669-2685, 2011
Proteins, Lipids, and Water in the Gas Phase
Part of Macromolecular Bioscience, p. 50-59, 2011
Part of Biophysical Journal, p. 1345-1353, 2011
Subunit Interface Dynamics in Hexadecameric Rubisco
Part of Journal of Molecular Biology, p. 1083-1098, 2011
On the Feasibility of Nanocrystal Imaging Using Intense and Ultrashort X-ray Pulses
Part of ACS Nano, p. 139-146, 2011
Part of Peptides, p. 553-559, 2011
Toward prediction of functional protein pockets using blind docking and pocket search algorithms
Part of Protein Science, p. 880-893, 2011
Atomistic simulation of ion solvation in water explains surface preference of halides
Part of Proceedings of the National Academy of Sciences of the United States of America, p. 6838-6842, 2011
Scrutinizing Molecular Mechanics Force Fields on the Submicrosecond Timescale with NMR Data
Part of Biophysical Journal, p. 647-655, 2010
Part of Journal of Chemical Theory and Computation, p. 3713-3720, 2010
Hawk: the image reconstruction package for coherent X-ray diffractive imaging
Part of Journal of applied crystallography, p. 1535-1539, 2010
Molecular Dynamics Simulations of a Membrane Protein-Micelle Complex in Vacuo
Part of Journal of the American Chemical Society, p. 16606-16607, 2009
Part of Physical Review E. Statistical, Nonlinear, and Soft Matter Physics, p. 31905, 2009
Encapsulation of myoglobin in a cetyl trimethylammonium bromide micelle in vacuo: a simulation study
Part of Biochemistry, p. 1006-1015, 2009
A density functional study of N-15 chemical shielding tensors in quinolines
Part of Chemical Physics Letters, p. 196-200, 2009
Structural stability of electrosprayed proteins: temperature and hydration effects
Part of Physical Chemistry, Chemical Physics - PCCP, p. 8069-8078, 2009
Solution conformations of an insect neuropeptide: crustacean cardioactive peptide (CCAP)
Part of Peptides, p. 557-564, 2009
Role of spin state on the geometry and nuclear quadrupole resonance parameters in hemin complex
Part of Biophysical Chemistry, p. 200-206, 2008
GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation
Part of Journal of Chemical Theory and Computation, p. 435-447, 2008
People
David van der Spoel, Ph. D., Professor
Marie-Madeleine Walz, Ph.D., Postdoc
Alfred Andersson, Ph.D. student
Kristian Kriz, Ph.D., Postdoc
Paul J. van Maaren, M.Sc. Visiting scientist
Contact
- www.folding.bmc.uu.se