van der Spoel labb
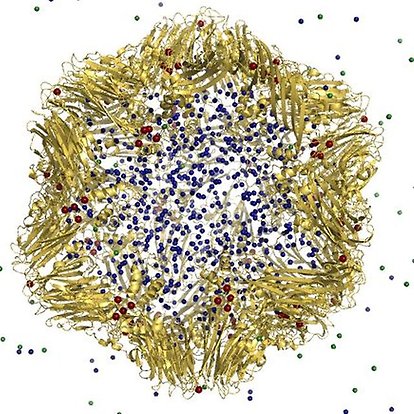
Min grupp arbetar på att förbättra modeller och metoder för att göra molekyldynamik (MD) simuleringar. Med MD simulering som baseras på en fysikalisk modell kan man studera hur molekyler interagerar och hur de rör sig i tiden. Resultaten kan analyseras för att ta fram grundläggande information om molekyler, vätskor och komplexa biologiska system såsom hela viruspartiklar. Vi studerar viruspartiklar i syfte att förstå infektion: vad händer när en viruspartikel kommer in i en cell? Hur kommer RNA:t ut ur kapsiden? Ett annat ämne vi arbetar på är att förstå hur RNA-genomet kan veckas inuti en liten viruspartikel. Slutligen forskar vi på hur nybyggda virus kan ta sig ut ur en cell igen.
Populärvetenskaplig presentation
Virus orsaker stora problem för växter och djur, och därför är det viktigt att förstå deras uppbyggnad, livscykler och hur infektionsprocessen går tillväga. Det finns en hel del mätningar kring virus, men vissa saker, såsom hur genomet inkapslas är svårt att ta reda på. Vi kommer att använda datasimuleringar för att bygga en komplett modell av en viruspartikel med genom, för att sedan kunna göra läkemedel. Bakterier som har blivit resistenta mot antibiotika är likaledes ett växande problem orsakat av överanvändning av läkemedel inom främst djurhållningen; till exempel används 80% av antibiotika i USA i djuruppfödningen och även om inte detta händer på samma skala i Sverige sprider sig de resistenta bakterier världen över. Det finns en klass naturliga peptider (en sammansättning av ett mindre antal aminosyror) med antibiotiska egenskaper. Här ska vi studera några sådana peptider som förmodas kan förstöra en cellvägg genom att bygga en kanal där vatten och andra små molekyler eller joner kan flytta igenom. På grund av denna egenskap kan sådana peptider bara användas utvärtes, t.ex. i ögonen eller på infekterade sår. Vi ska mäta om det är rimligt att sådana kanaler byggs utav dessa peptider och hur snabbt vatten och joner kan transporteras igenom kanalerna. Inom min forskning används simuleringar med hjälp av datamodeller som har byggts och förfinats under decennier. Även om dessa modeller är ganska bra finns det mycket utrymme för förbättring. Här ska vi även arbeta på att göra bättre modeller och verktyg för sådana simuleringar. Dessa verktyg kommer att distribueras som öppen källkod för att andra forskare inom universitetsvärlden eller industri kan granska koden. De nya modellerna kommer att förfinas och testas på organiska vätskor. Noggranna modeller för sådana vätskor är av stort intresse för kemiindustrin. I och med dessa nya metoder samt de avancerade tillämpningar kommer det här projektet att flytta fram kunskapsnivån inom många områden.
Forskning
...
Välkommen till vår extern hemsida
Gruppmedlemmar
Publikationer
Martini on the Rocks: Can a Coarse-Grained Force Field Model Crystals?
Ingår i Journal of Physical Chemistry Letters, s. 1079-1088, 2024
- DOI för Martini on the Rocks: Can a Coarse-Grained Force Field Model Crystals?
- Ladda ner fulltext (pdf) av Martini on the Rocks: Can a Coarse-Grained Force Field Model Crystals?

Ingår i Journal of Chemical Theory and Computation, s. 2362-2376, 2024
- DOI för Impact of Combination Rules, Level of Theory, and Potential Function on the Modeling of Gas- and Condensed-Phase Properties of Noble Gases
- Ladda ner fulltext (pdf) av Impact of Combination Rules, Level of Theory, and Potential Function on the Modeling of Gas- and Condensed-Phase Properties of Noble Gases
Can molecular dynamics be used to simulate biomolecular recognition?
Ingår i Journal of Chemical Physics, 2023
Simulations of Amyloid-Forming Peptides in the Crystal State
Ingår i The Protein Journal, s. 192-204, 2023
- DOI för Simulations of Amyloid-Forming Peptides in the Crystal State
- Ladda ner fulltext (pdf) av Simulations of Amyloid-Forming Peptides in the Crystal State
Probing Phase Transitions in Organic Crystals Using Atomistic MD Simulations
Ingår i ACS PHYSICAL CHEMISTRY AU, s. 84-93, 2023
- DOI för Probing Phase Transitions in Organic Crystals Using Atomistic MD Simulations
- Ladda ner fulltext (pdf) av Probing Phase Transitions in Organic Crystals Using Atomistic MD Simulations
Binding Networks Identify Targetable Protein Pockets for Mechanism-Based Drug Design
Ingår i International Journal of Molecular Sciences, 2022
- DOI för Binding Networks Identify Targetable Protein Pockets for Mechanism-Based Drug Design
- Ladda ner fulltext (pdf) av Binding Networks Identify Targetable Protein Pockets for Mechanism-Based Drug Design
Editorial overview: Theory and simulation and their new friends
Ingår i Current opinion in structural biology, 2021
Ingår i Journal of Chemical Physics, 2021
Ingår i Journal of Chemical Physics, 2021
Accurate absolute free energies for ligand-protein binding based on non-equilibrium approaches
Ingår i Communications Chemistry, 2021
- DOI för Accurate absolute free energies for ligand-protein binding based on non-equilibrium approaches
- Ladda ner fulltext (pdf) av Accurate absolute free energies for ligand-protein binding based on non-equilibrium approaches
Microscopic origins of conductivity in molten salts unraveled by computer simulations
Ingår i Communications Chemistry, 2021
- DOI för Microscopic origins of conductivity in molten salts unraveled by computer simulations
- Ladda ner fulltext (pdf) av Microscopic origins of conductivity in molten salts unraveled by computer simulations
NMR Refinement and Peptide Folding Using the GROMACS Software
Ingår i Journal of Biomolecular NMR, s. 143-149, 2021
- DOI för NMR Refinement and Peptide Folding Using the GROMACS Software
- Ladda ner fulltext (pdf) av NMR Refinement and Peptide Folding Using the GROMACS Software
Making Soup: Preparing and Validating Models of the Bacterial Cytoplasm for Molecular Simulation
Ingår i Journal of Chemical Information and Modeling, s. 322-331, 2020
Theoretical Infrared Spectra: Quantitative Similarity Measures and Force Fields
Ingår i Journal of Chemical Theory and Computation, s. 3307-3315, 2020
- DOI för Theoretical Infrared Spectra: Quantitative Similarity Measures and Force Fields
- Ladda ner fulltext (pdf) av Theoretical Infrared Spectra: Quantitative Similarity Measures and Force Fields
An Intuitively Understandable Quality Measure for Theoretical Vibrational Spectra
Ingår i Journal of Physical Chemistry Letters, s. 5471-5475, 2020
- DOI för An Intuitively Understandable Quality Measure for Theoretical Vibrational Spectra
- Ladda ner fulltext (pdf) av An Intuitively Understandable Quality Measure for Theoretical Vibrational Spectra
A potential for molecular simulation of compounds with linear moieties
Ingår i Journal of Chemical Physics, 2020
- DOI för A potential for molecular simulation of compounds with linear moieties
- Ladda ner fulltext (pdf) av A potential for molecular simulation of compounds with linear moieties
Toward a Computational Ecotoxicity Assay
Ingår i Journal of Chemical Information and Modeling, s. 3792-3803, 2020
Propagation of uncertainty in physicochemical data to force field predictions
Ingår i Physical Review Research, 2020
- DOI för Propagation of uncertainty in physicochemical data to force field predictions
- Ladda ner fulltext (pdf) av Propagation of uncertainty in physicochemical data to force field predictions
Ingår i PloS Computational Biology, 2019
- DOI för Ten simple rules on how to create open access and reproducible molecular simulations of biological systems
- Ladda ner fulltext (pdf) av Ten simple rules on how to create open access and reproducible molecular simulations of biological systems
Role of Host-Guest Charge Transfer in Cyclodextrin Complexation: A Computational Study
Ingår i The Journal of Physical Chemistry C, s. 17745-17756, 2019
Molten alkali halides - temperature dependence of structure, dynamics and thermodynamics
Ingår i Physical Chemistry, Chemical Physics - PCCP, s. 18516-18524, 2019
Direct Link between Structure, Dynamics, and Thermodynamics in Molten Salts
Ingår i The Journal of Physical Chemistry C, s. 25596-25602, 2019
Systematically improved melting point prediction: a detailed physical simulation model is required
Ingår i Chemical Communications, s. 12044-12047, 2019
- DOI för Systematically improved melting point prediction: a detailed physical simulation model is required
- Ladda ner fulltext (pdf) av Systematically improved melting point prediction: a detailed physical simulation model is required
Rotational and Translational Diffusion of Proteins as a Function of Concentration
Ingår i ACS Omega, s. 20654-20664, 2019
- DOI för Rotational and Translational Diffusion of Proteins as a Function of Concentration
- Ladda ner fulltext (pdf) av Rotational and Translational Diffusion of Proteins as a Function of Concentration
Prediction of Partition Coefficients of Environmental Toxins Using Computational Chemistry Methods
Ingår i ACS Omega, s. 13772-13781, 2019
- DOI för Prediction of Partition Coefficients of Environmental Toxins Using Computational Chemistry Methods
- Ladda ner fulltext (pdf) av Prediction of Partition Coefficients of Environmental Toxins Using Computational Chemistry Methods
Transient isomers in the photodissociation of bromoiodomethane
Ingår i Journal of Chemical Physics, 2018
Force Field Benchmark of Amino Acids: I. Hydration and Diffusion in Different Water Models
Ingår i Journal of Chemical Information and Modeling, s. 1037-1052, 2018
Statistical efficiency of methods for computing free energy of hydration
Ingår i Journal of Chemical Physics, 2018
- DOI för Statistical efficiency of methods for computing free energy of hydration
- Ladda ner fulltext (pdf) av Statistical efficiency of methods for computing free energy of hydration
Impact of Dispersion Coefficient on Simulations of Proteins and Organic Liquids
Ingår i Journal of Physical Chemistry B, s. 8018-8027, 2018
Small Molecule Thermochemistry: A Tool for Empirical Force Field Development
Ingår i Journal of Physical Chemistry A, s. 8982-8988, 2018
Ingår i Scientific Data, 2018
- DOI för The Alexandria library, a quantum-chemical database of molecular properties for force field development
- Ladda ner fulltext (pdf) av The Alexandria library, a quantum-chemical database of molecular properties for force field development
Phase-Transferable Force Field for Alkali Halides
Ingår i Journal of Chemical Theory and Computation, s. 5933-5948, 2018
Ingår i Journal of Chemical Theory and Computation, s. 5553-5566, 2018
Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations
Ingår i Nucleic Acids Research, s. 4872-4882, 2018
- DOI för Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations
- Ladda ner fulltext (pdf) av Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations
Ingår i Journal of Physical Chemistry Letters, s. 2705-2712, 2017
Ingår i Journal of Chemical Theory and Computation, s. 1034-1043, 2017
Ingår i Journal of Physics, 2017
Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1
Ingår i Molecular Cell, s. 847-8590000000, 2017
- DOI för Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1
- Ladda ner fulltext (pdf) av Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1
Systematic exploration of multiple drug binding sites
Ingår i Journal of Cheminformatics, 2017
- DOI för Systematic exploration of multiple drug binding sites
- Ladda ner fulltext (pdf) av Systematic exploration of multiple drug binding sites
Ingår i Journal of Chemical Physics, 2016
- DOI för Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity
- Ladda ner fulltext (pdf) av Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity
Atomistic Simulation of Protein Encapsulation in Metal-Organic Frameworks
Ingår i Journal of Physical Chemistry B, s. 477-484, 2016
Exploration of Interfacial Hydration Networks of Target Ligand Complexes
Ingår i Journal of Chemical Information and Modeling, s. 148-158, 2016
Ingår i Journal of Chemical Information and Modeling, s. 2080-2092, 2016
- DOI för Evaluation of Generalized Born Models for Large Scale Affinity Prediction of Cyclodextrin Host-Guest Complexes
- Ladda ner fulltext (pdf) av Evaluation of Generalized Born Models for Large Scale Affinity Prediction of Cyclodextrin Host-Guest Complexes
Binding of Pollutants to Biomolecules: A Simulation Study
Ingår i Chemical Research in Toxicology, s. 1679-1688, 2016
- DOI för Binding of Pollutants to Biomolecules: A Simulation Study
- Ladda ner fulltext (pdf) av Binding of Pollutants to Biomolecules: A Simulation Study
Ingår i Journal of Chemical Physics, 2016
Ingår i Journal of Chemical Theory and Computation, s. 5103-5113, 2015
Properties of Organic Liquids when Simulated with Long-Range Lennard-Jones Interactions
Ingår i Journal of Chemical Theory and Computation, s. 2938-2944, 2015
Ingår i Journal of Computational Chemistry, s. 1473-1479, 2015
Ingår i Journal of Molecular Structure, s. 196-202, 2015
Mobility-based prediction of hydration structures of protein surfaces
Ingår i Bioinformatics, s. 1959-1965, 2015
Deciphering Solution Scattering Data with Experimentally Guided Molecular Dynamics Simulations
Ingår i Journal of Chemical Theory and Computation, s. 780-787, 2015
- DOI för Deciphering Solution Scattering Data with Experimentally Guided Molecular Dynamics Simulations
- Ladda ner fulltext (pdf) av Deciphering Solution Scattering Data with Experimentally Guided Molecular Dynamics Simulations
Force Field Benchmark of Organic Liquids. 2. Gibbs Energy of Solvation
Ingår i JOURNAL OF CHEMICAL INFORMATION AND MODELING, s. 1192-1201, 2015
Ingår i PLOS ONE, 2015
- DOI för Deconvoluting Protein (Un)folding Structural Ensembles Using X-Ray Scattering, Nuclear Magnetic Resonance Spectroscopy and Molecular Dynamics Simulation
- Ladda ner fulltext (pdf) av Deconvoluting Protein (Un)folding Structural Ensembles Using X-Ray Scattering, Nuclear Magnetic Resonance Spectroscopy and Molecular Dynamics Simulation
Thermodynamics of hydronium and hydroxide surface solvation
Ingår i Chemical Science, s. 1745-1749, 2014
Ingår i Journal of Chemical Theory and Computation, s. 5606-5615, 2014
Ingår i The Journal of Physical Chemistry C, s. 7163-7173, 2014
CO2 and O-2 Distribution in Rubisco Suggests the Small Subunit Functions as a CO2 Reservoir
Ingår i Journal of the American Chemical Society, s. 3165-3171, 2014
Thiamin Function, Metabolism, Uptake, and Transport
Ingår i Biochemistry, s. 821-835, 2014
Ingår i Bioinformatics, s. 439-441, 2014
Ingår i Journal of Computational Chemistry, s. 260-269, 2014
Local Partition Coefficients Govern Solute Permeability of Cholesterol-Containing Membranes
Ingår i Biophysical Journal, s. 2760-2770, 2013
Transcription-factor binding and sliding on DNA studied using micro- and macroscopic models
Ingår i Proceedings of the National Academy of Sciences of the United States of America, s. 19796-19801, 2013
Ingår i Environmental Science and Technology, s. 7421-7429, 2013
Ingår i Peptides, s. 94-100, 2013
GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit
Ingår i Bioinformatics, s. 845-854, 2013
Unexpected Effects of Cholesterol on Membrane Permeability
Ingår i Biophysical Journal, 2013
Ingår i Biochemistry, s. 7798-7806, 2013
Quantification of Solvent Contribution to the Stability of Noncovalent Complexes
Ingår i Journal of Chemical Theory and Computation, s. 4542-4551, 2013
Organic molecules on the surface of water droplets: an energetic perspective
Ingår i Physical Chemistry, Chemical Physics - PCCP, s. 9537-9545, 2012
GROMACS molecule & liquid database
Ingår i Bioinformatics, s. 752-753, 2012
Ingår i Journal of the American Society for Mass Spectrometry, s. 1319-1325, 2012
Molecular recognition in different environments: β-Cyclodextrin dimer formation in organic solvents
Ingår i Journal of Physical Chemistry B, s. 12684-12693, 2012
Free Energy of Separation of Structure II Clathrate Hydrate in Water and a Light Oil
Ingår i Journal of Physical Chemistry B, s. 5933-5940, 2012
Ingår i Journal of Chemical Theory and Computation, s. 2474-2483, 2012
Virus Capsid Dissolution Studied by Microsecond Molecular Dynamics Simulations
Ingår i PloS Computational Biology, 2012
- DOI för Virus Capsid Dissolution Studied by Microsecond Molecular Dynamics Simulations
- Ladda ner fulltext (pdf) av Virus Capsid Dissolution Studied by Microsecond Molecular Dynamics Simulations
Large Influence of Cholesterol on Solute Partitioning into Lipid Membranes
Ingår i Journal of the American Chemical Society, s. 5351-5361, 2012
Ingår i Journal of Chemical Theory and Computation, s. 61-74, 2012
Ingår i Journal of Physical Chemistry B, s. 3880-3889, 2012
Ingår i Wiley Interdisciplinary Reviews: Computational Molecular Science, s. 710-715, 2011
Ingår i Physical Review E. Statistical, Nonlinear, and Soft Matter Physics, 2011
Ingår i Journal of Molecular Modeling, s. 3289-3297, 2011
Trajectory NG: portable, compressed, general molecular dynamics trajectories
Ingår i Journal of Molecular Modeling, s. 2669-2685, 2011
Proteins, Lipids, and Water in the Gas Phase
Ingår i Macromolecular Bioscience, s. 50-59, 2011
Ingår i Biophysical Journal, s. 1345-1353, 2011
Subunit Interface Dynamics in Hexadecameric Rubisco
Ingår i Journal of Molecular Biology, s. 1083-1098, 2011
On the Feasibility of Nanocrystal Imaging Using Intense and Ultrashort X-ray Pulses
Ingår i ACS Nano, s. 139-146, 2011
Ingår i Peptides, s. 553-559, 2011
Toward prediction of functional protein pockets using blind docking and pocket search algorithms
Ingår i Protein Science, s. 880-893, 2011
Atomistic simulation of ion solvation in water explains surface preference of halides
Ingår i Proceedings of the National Academy of Sciences of the United States of America, s. 6838-6842, 2011
Scrutinizing Molecular Mechanics Force Fields on the Submicrosecond Timescale with NMR Data
Ingår i Biophysical Journal, s. 647-655, 2010
Ingår i Journal of Chemical Theory and Computation, s. 3713-3720, 2010
Hawk: the image reconstruction package for coherent X-ray diffractive imaging
Ingår i Journal of applied crystallography, s. 1535-1539, 2010
Molecular Dynamics Simulations of a Membrane Protein-Micelle Complex in Vacuo
Ingår i Journal of the American Chemical Society, s. 16606-16607, 2009
Ingår i Physical Review E. Statistical, Nonlinear, and Soft Matter Physics, s. 31905, 2009
Encapsulation of myoglobin in a cetyl trimethylammonium bromide micelle in vacuo: a simulation study
Ingår i Biochemistry, s. 1006-1015, 2009
A density functional study of N-15 chemical shielding tensors in quinolines
Ingår i Chemical Physics Letters, s. 196-200, 2009
Structural stability of electrosprayed proteins: temperature and hydration effects
Ingår i Physical Chemistry, Chemical Physics - PCCP, s. 8069-8078, 2009
Solution conformations of an insect neuropeptide: crustacean cardioactive peptide (CCAP)
Ingår i Peptides, s. 557-564, 2009
Role of spin state on the geometry and nuclear quadrupole resonance parameters in hemin complex
Ingår i Biophysical Chemistry, s. 200-206, 2008
GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation
Ingår i Journal of Chemical Theory and Computation, s. 435-447, 2008
Medarbetare
David van der Spoel, Ph. D., Professor
Marie-Madeleine Walz, Ph.D., Postdoc
Alfred Andersson, Ph.D. student
Kristian Kriz, Ph.D., Postdoc
Paul J. van Maaren, M.Sc. Visiting scientist